Ortholuge and OrtholugeDB
Publications
The OrtholugeDB database is described in the paper:
The Ortholuge method is described in the paper:
The method for computing the local False Discovery Rate for the Ortholuge ratios is described in the paper:
Ortholuge
Ortholuge is a computational method that can generate precise ortholog predictions between two species on a genome-wide scale (using additional outgroup genome for reference). It computes phylogenetic distance ratios for each pair of orthologs that reflect the relative rate of divergence for the orthologs (figure 1).
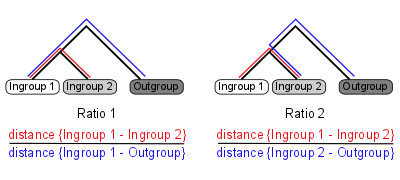
Figure 1: Ortholuge phylogenetic ratios
Two ratios are needed to summarize the relative branch lengths of both ingroup genes. These phylogenetic ratios allow you to distinguish between predicted orthologs with phylogenetic distance that is comparable to the species divergence (termed SSD orthologs: supporting-species-divergence orthologs) and predicted orthologs with unusual divergence (Non-SSD). Unusual divergence is observable when the ratios are plotted on a genome-wide scale (figure 2).
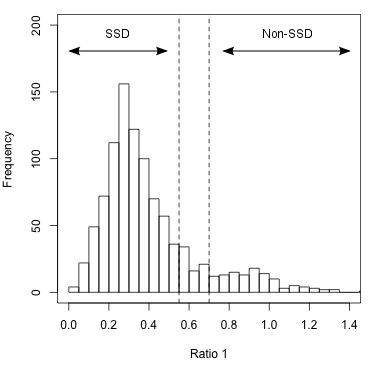
Figure 2: Histogram of Ratio 1 values for entire genome
Ortholog and Inparalog Prediction
In OrtholugeDB, the reciprocal-best-BLAST-hit (RBBH) procedure is used to generate the initial set of ortholog predictions. Genes are declared orthologs if they are each other's top BLAST hit when each genome is BLAST'ed against the other. The top hit is determined by having the lowest e-value and highest bit score. Multiple RBBHs are possible, and we keep track of all of them (these cases often represent very recent gene duplications). We evaluate the RBBH-predicted orthologs using Ortholuge.
Inparalogs are ortholog genes that have duplicated (subsequent to the species divergence). If the genes duplicated prior to the speciation (and creation of orthologs) the genes are referred to as outparalogs. We identify inparalogs using a procedure based on the InParanoid method, a gene is declared an inparalog if its BLAST bit-score to the ortholog gene from the same species is higher than the score between the orthologous genes from different species. We also check that the inparalog's top BLAST hit in the other species is the same as its ortholog partner from the same species. Inparanoid is described in this paper:
Ostlund G, Schmitt T, Forslund K, Kostler T, Messina DN, Roopra S, Frings O and Sonnhammer ELL. InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Research, 2009. 38:D196-D203.
Classifications
The following Ortholuge classifications are used in OrtholugeDB:
| SSD |
(Supporting species divergence). Predicted orthologs whose divergence (as reported by the Ortholuge phylogenetic ratios) is consistent with the divergence observed for the species. These predicted orthologs likely represent valid orthologs. |
| Borderline-SSD |
Predicted orthologs with a phylogenetic ratio that is slightly higher than expected. If precision is important to your application you may want to exclude these orthologs. |
| Divergent Non-SSD |
Non-SSD genes have phylogenetic ratios that are significantly higher when compared to other orthologs in the genomes, indicating that their divergence is not consistent with the species level of divergence. Divergent Non-SSD genes are diverging atypically for the genomes and also have a significant phylogenetic distance separating them. These are often incorrectly-predicted orthologs, or orthologs that have undergone unusual phylogenetic divergence. |
| Similar Non-SSD |
Non-SSD genes have phylogenetic ratios that are significantly higher when compared to other orthologs in the genomes, indicating that their divergence is not consistent with the species level of divergence. Similar Non-SSD genes have diverged unusually; the length of one of the branches in the gene tree is proportionally longer than expected, however the total phylogenetic distance separating the predicted orthologs is relatively small. Many Similar Non-SSD genes will often be valid orthologs. The high phylogenetic ratio may suggest the genes are evolving at different rates. |
| RBB |
(Reciprocal Best-BLAST). Orthologs predicted by reciprocal-best-BLAST analysis that have not undergone analysis by Ortholuge. |
Boundaries between the Ortholuge classifications are determined by the ratios and their corresponding false discovery rates (FDR). Our method for computing a local FDR for a given ratio value is described here:
Min JE, Whiteside MD, Brinkman FSL, McNeney B, Graham J. A statistical approach to high-throughput screening of predicted orthologs. Computational Statistics and Data Analysis, 2011. 55(1): 953-43.
This local FDR approach is an extension of the Ortholuge method first described here:
Fulton DL, Li YY, Laird M, Horsman BGS, Roche FMR, Brinkman FSL. Improving the specificity of high-throughput ortholog prediction. BMC Bioinformatics, 2006. 7:270.
Outgroups
Ortholuge uses an outgroup genome as a reference for computing the phylogenetic distance ratios. In OrtholugeDB, outgroups are automatically selected based on their CVtree distance from the ingroups (we predetermined the optimal CVtree distance for the outgroup provided with CVtree distance for the ingroups). CVtree distances are a composition-based distance metric that reflect the evolutionary relatedness between species proteomes. The following paper describes the CVtree tool:
Xu Z, Hao B. CVTree update: a newly designed phylogenetic study platform using composition vectors and whole genomes. Nucleic Acids Research, 2009. 37(Web Server issue):W174-8
Software
The Ortholuge software is available for download from the following site:
http://www.pathogenomics.ca/ortholuge/
OrtholugeDB
OrtholugeDB is a comprehensive database of bacterial and archaeal orthologs. This database provides Ortholuge-based ortholog predictions for all fully-sequenced bacteria and archaea genomes where a suitable outgroup is available and RBBH predictions otherwise. Ortholog predictions are available for protein-coding genes only. Data is stored in a MySQL database. We will provide a dump of the database upon request.
Web Interface
We provide the following types of searches to retrieve data from OrtholugeDB:
-
Obtain Orthologs Between Two GenomesObtain all orthologs between two genomes. Ortholuge results will be displayed in a separate column when available (Analysis Type: at the top of the page will tell you whether Ortholuge or Reciprocal Best BLAST analysis was performed). Alternatively, you can return genes in one of the species that do not have orthologs (and are not inparalogs) by selecting a genome in the Optional - only return unique genes for: form.
-
Obtain Orthologs For a Single Gene
Obtain all orthologs for a gene. Search in all genomes or restrict search to a specific set of genomes. You will be required to enter the gene's Entrez GeneID, GI number, locus tag, Refseq accession or gene/product name. Selecting Show gene context: will show an image of the gene neighbourhood surrounding the ortholog genes.
-
Compare Reference Genome to Orthologs in Comparison Genomes
Obtain orthologs between a single genome of interest (reference genome) and one or more other genomes (comparison genomes).
This is a 2-step search. For step 1, choose a reference genome and genomes to compare. In step 2, the comparison genomes can be used to filter the genes in the reference genome. The criteria are:
Ortholog is Present Genes in the reference genome that have orthologs in comparison species are displayed.
Ortholog is Absent Genes in the reference genome that do not have orthologs in comparison species are displayed.
Ortholog is Optional Genes in the reference genome are displayed regardless if they have orthologs in this comparison genome or not. In other words, the comparison genome is not used to filter but its orthologs are still displayed.
Multiple criteria are combined with logical AND operator (i.e. genes for which all conditions are true are returned). This filtering function allows you identify genes that, for example, have orthologs in species x and y but not z.
The search returns a phyletic matrix that gives a high-level view of the genes in your reference that have or are lacking orthologs, and if they have orthologs, are there multiple co-orthologs or inparalogs. Five codes are used to describe the ortholog cardinality:
0 Gene in reference genome has no ortholog in comparison genome.
1:1 Gene has one ortholog in the comparison genome. No inparalogs/co-orthologs were identified.
1:M Gene has ortholog in the comparison genome. One or more inparalogs/co-orthologs were identified for the ortholog gene in comparison genome (suggesting duplication of the orthologous gene after speciation).
M:1 Gene has ortholog in the comparison genome. One or more inparalogs/co-orthologs were identified for the ortholog gene in reference genome (suggesting duplication of the orthologous gene after speciation).
M:M One or more inparalogs/co-orthologs were identified for the orthologous genes in both the comparison and reference genomes (suggesting tandem duplications in both species after species divergence).
The coloring scheme of the cells in the matrix are used to convey the Ortholuge classifications of the orthologs
SSD 
Borderline 
Similar Non-SSD 
Divergent Non-SSD 
RBB 
More information on the Ortholuge classifications is available here.
Gene duplications can impact how gene function evolves in species. Knowledge of duplication events, combined with the ortholog divergence information provided by the Ortholuge ratios, can help inform you regarding which predicted orthologs have likely similar functions and which have divergent functions.
In the result pages for these searches, the number of inparalogs associated with an ortholog will be listed in the Inparalog column. Clicking on the link in this column will take you to a more detailed view that will list the individual inparalogs and orthologs associated with the genes in that row, along with the ortholog's ratios values and local FDR rates. We have computed the Ortholuge data for inparalogs as well and this data will also be available here.
In this view, orthologs are sets of genes that are reciprocal best BLAST hits of each other. Using this operational definition, multiple RBB orthologs can be identified. Inparalogs are identified using procedures based on the Inparanoid method. If there is no available outgroup ortholog, only reciprocal best BLAST predictions will be available and the column will contain RBB. Further details are available here.
Data Sources
The protein sequences for completely sequenced bacteria and archaea species are obtained through the MicrobeDB resource. MicrobeDB is described in the following paper:
Langille MG, Laird MR, Hsiao WW, Chiu TA, Eisen JA, Brinkman FS. MicrobeDB: a locally maintainable database of microbial genomic sequences. Bioinformatics. Bioinformatics. 2012 Jul 15;28(14):1947-8.
Downloading
Data can be downloaded from a results page in the following formats:
- Comma-separated values (CSV)
- Tab-delimited
- OrthoXML. The xml specification is available from here: seqxml.org.
Data is housed in a MySQL database. We will also provide a MySQL dump of the database upon request.
Contact
OrtholugeDB is developed by the Brinkman lab at Simon Fraser University, Burnaby (Greater Vancouver), BC, Canada.
For help or feedback, please email us at ortholugedb-mail@sfu.ca.
Acknowledgements
OrtholugeDB development has been funded by Genome Canada/Genome BC with the support of Cystic Fibrosis Foundation Therapeutics Inc..